
Rのggplotを使ってきれいなグラフを作りたいという方は多いのではないでしょうか。
この記事ではRのggplotを始めるための基本的な知識をまとめています。
本記事を通して、ggplotを扱うための基本的な方法を学びましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
作画ツールggplotとは?
ggplotは、R言語において美しく柔軟なグラフを描画するためのパッケージです。各種グラフのタイプ(散布図、ヒストグラム、箱ひげ図など)もggplotを使用することで描画することができます。生命科学において、RNA-seq解析の結果を視覚化するために使用されます。RNA-seq解析によって得られた遺伝子発現量のデータを可視化することで、遺伝子の発現パターンを理解することができます。美しく柔軟なグラフを簡単に描画することができるのでR言語を使う醍醐味だと考える人も多いです。
Rstudioの設定
R環境の用意
Rを実行するにははじめに環境構築が必要となります。今回はRを実行する環境としてRstudioを使っていきます。
各OS環境を元にこちらのサイトよりインストーラーをダウンロードしてください。https://cran.r-project.org/
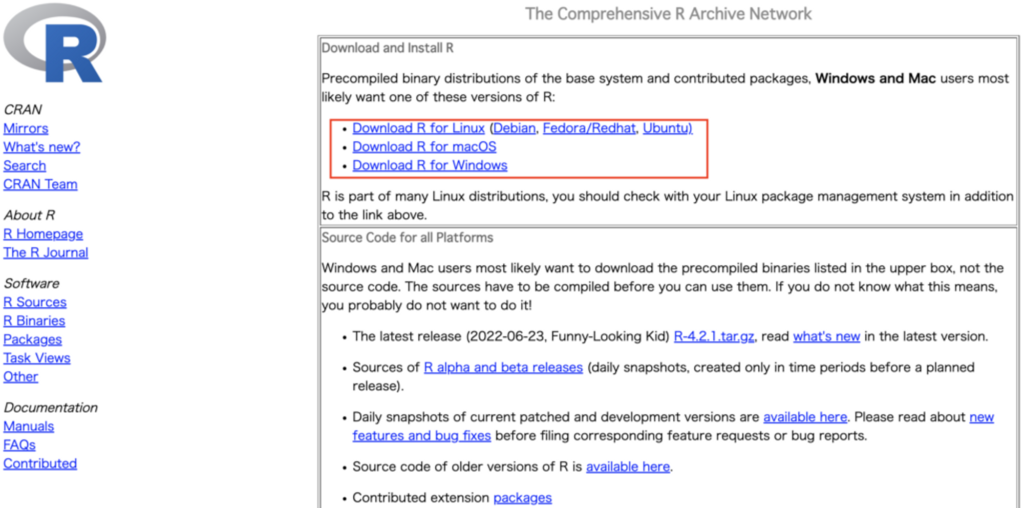
インストールするバージョンは最新版を選んでください。一番トップにあるものが最新版です。

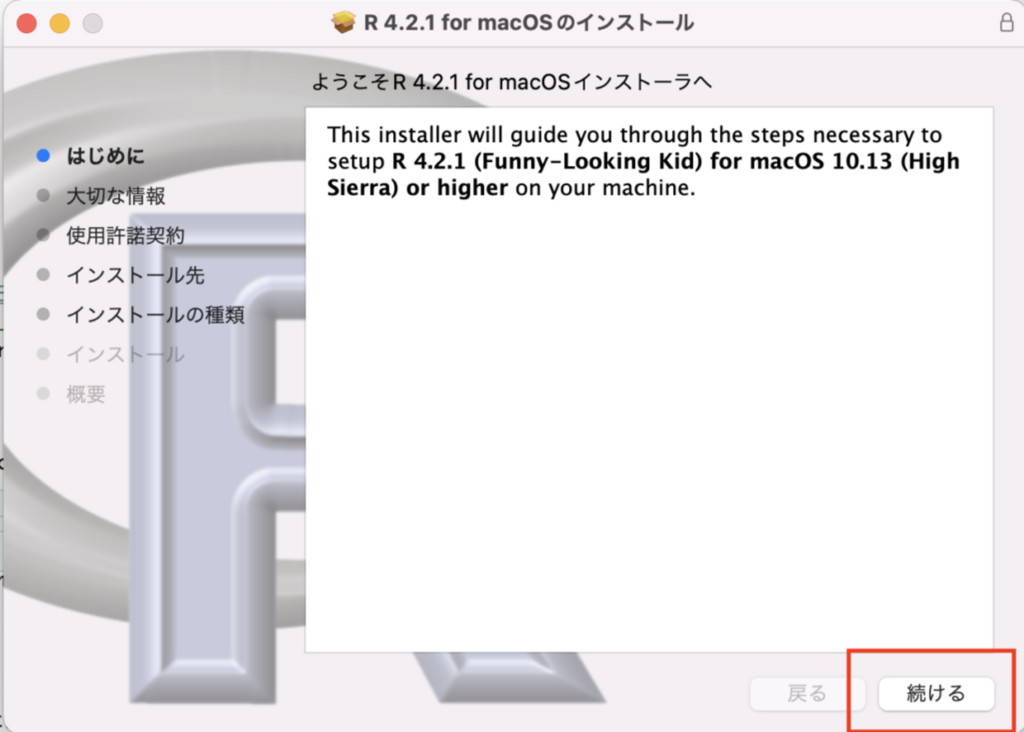
インストール画面では「続ける」を押して完了させてください。
Rstudioでは左上のエリアにコードを書いていきます。Runを押すと行実行ができます。Sourceを押すとスクリプト全体が実行できますので覚えておいてください。
ggplotの基本的な文法
今回は遺伝子発現量を棒グラフにするやり方をベースに記述します。ライブラリを読み込んでいきます。
install.packages("ggplot2")
library("ggplot2")今回使うデータに関しては以下になります。自分のサンプルでやる時はGeneとRatioの中身を変更して使用してください。
gene_ratio <- data.frame(
Gene = c("IL2", "IL4", "IL6", "IL8", "IL10", "TNF", "IFNG", "CD4", "CD8A", "CD19"),
Ratio = c(2.34, 5.67, 12.34, 1.11, 3.31, 5.76, 9.11, 4.30, 6.76, 2.22)
)まずは、棒グラフを作成するこちらのコードが基本となります。ggplot 関数を使っていきます。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) + geom_bar(stat="identity")以下のようなグラフを出力できれば成功です!
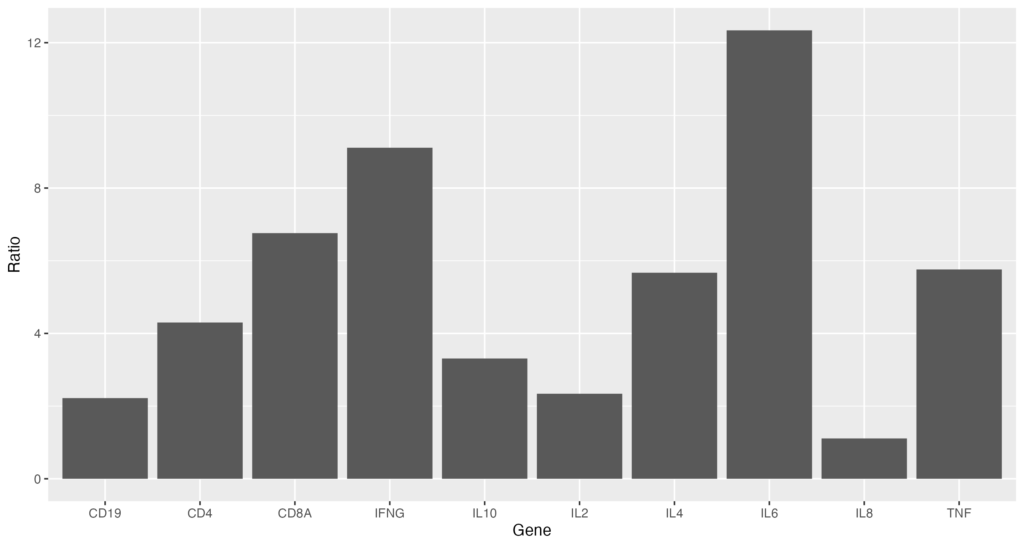
data として先程読み込んだgene_ratioを使います。aes 関数でx、y軸の情報を与えます。ここでは、x軸に Gene、y軸に Ratio を割り当てています。
次に、 geom_bar 関数を使用して、棒グラフを作成します。 stat="identity" を指定することで、各バーの高さが Ratio の値と一致するようにしています。
ではこちらのグラフをもとにグラフ描画を変えてみましょう。まずは軸のフォントサイズが小さいので大きくしてみます。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
theme(axis.text=element_text(size=15))以下のようなグラフを出力されます。軸のフォントサイズが大きくなりました!
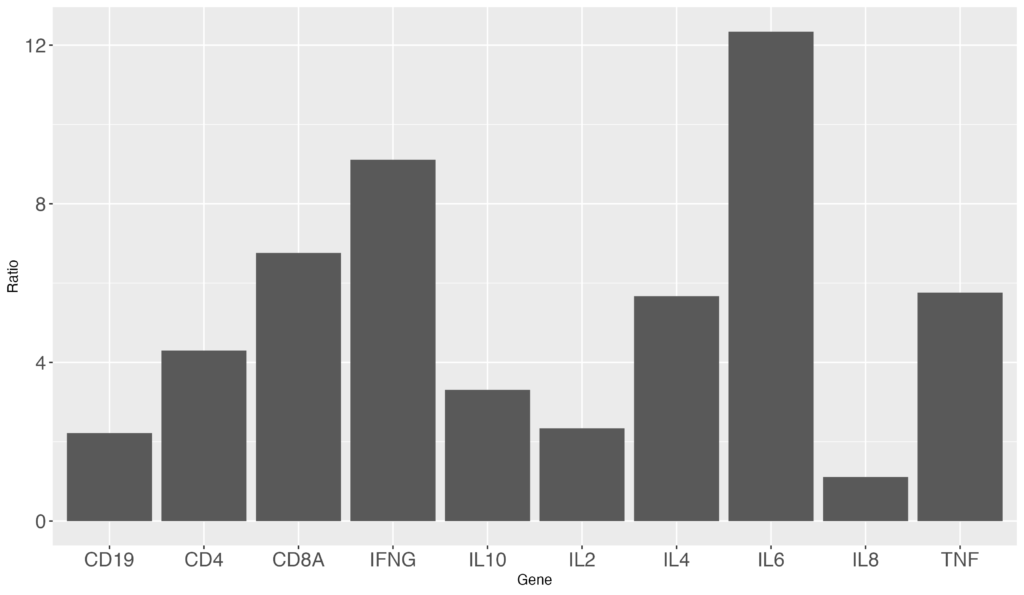
+ theme(axis.text=element_text(size=15)) を追加しました。+を忘れないように注意してください。
ちなみにaxis.textの部分をaxis.text.x のように指定すると、x軸だけ大きくすることも可能です。
y軸を大きくしたい場合は、axis.text.y を指定してください。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
theme(axis.text.x=element_text(size=15))
次はラベルのフォントサイズ大きくしてみましょう。theme(axis.title=element_text(size=20)) を追記します。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
theme(axis.title=element_text(size=20))ラベルのフォントサイズを20に調整してみました。
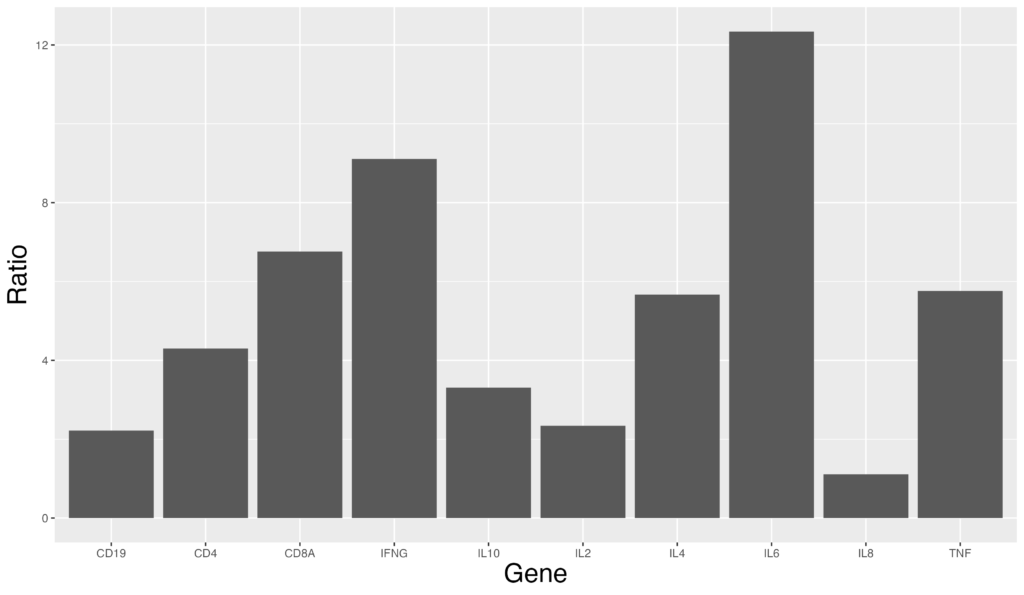
棒グラフでは発現量順に並び替えたいという要望もあると思います。その場合は降順ソートを用います。以下のコードで実現可能です。
ggplot(data = gene_ratio, aes(x = reorder(Gene, -Ratio), y = Ratio)) +
geom_bar(stat = "identity")左から降順に並び替えられます。
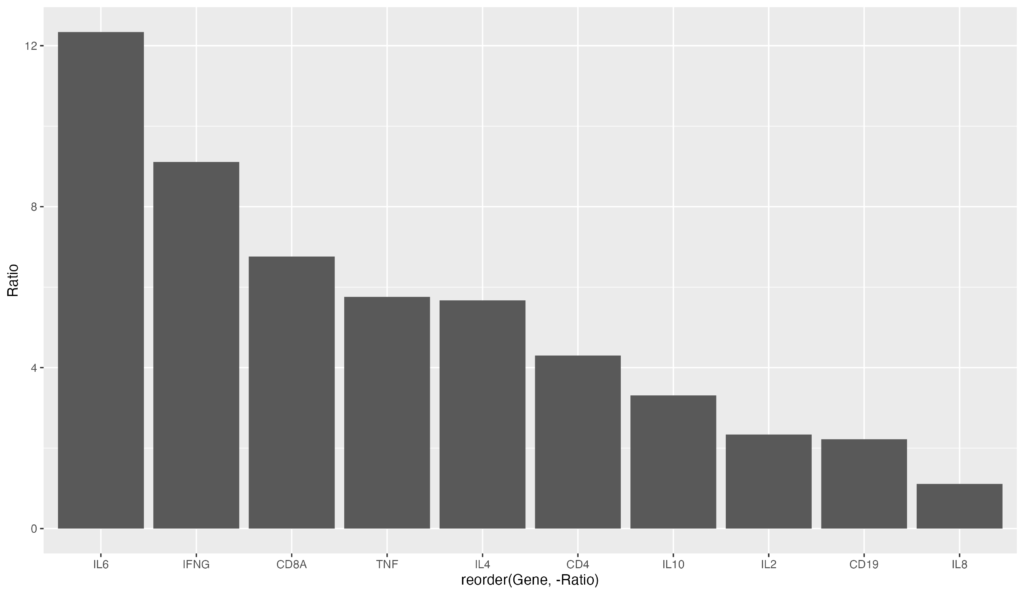
逆に、昇順にソートする場合は以下のコードになります。reorder(Gene, Ratio) でRatioのマイナスを削除するだけです。
ggplot(data = gene_ratio, aes(x = reorder(Gene, Ratio), y = Ratio)) +
geom_bar(stat = "identity")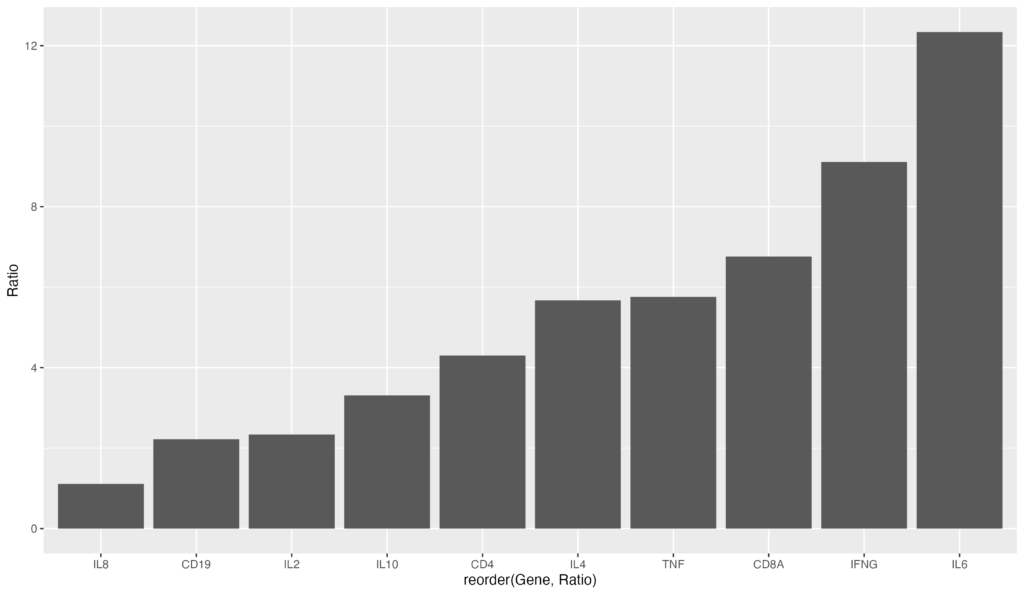
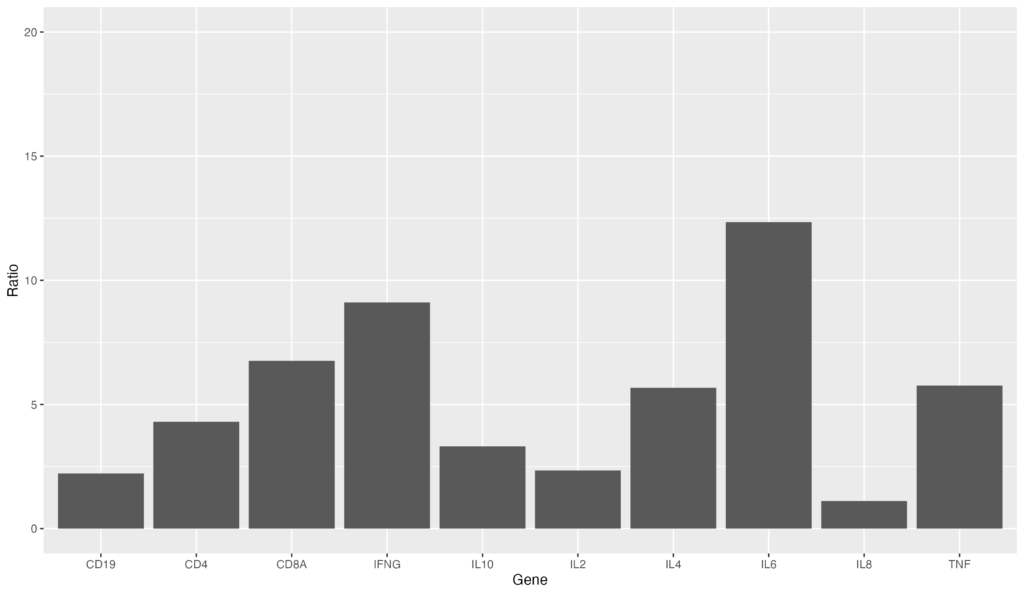
ggplotでは縦軸を自動調整してくれますが、自分の設定した任意の値にすることが出来ます。ylim(0, 20)を設定してみましょう。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat = "identity") +
ylim(0, 20)縦軸のMax値が20になります。
グラフタイトルをつけるには、labs(title = "Gene Ratio") のように指定します。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
labs(title = "Gene Ratio")
グラフタイトルを中央に持ってきたいときはtheme(plot.title = element_text(hjust = 0.5)) で指定しましょう。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
labs(title = "Gene Ratio") +
theme(plot.title = element_text(hjust = 0.5))
ラベル名を任意の値に変更したいときもあると思います。 xlab("GeneName") のように指定することで変更できます。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat = "identity") +
xlab("GeneName")
次に、barの色を変更してみましょう。 fill = "red" のようにfillの引数で色を割り当てます。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat = "identity", fill = "red")barを赤色にすることが出来ました。
ちなみに先程はfill=”red”としましたが、カラーコード自体を指定することが出来ます。こちらでカラーコードを探すことが出来ますのでみてみてください。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat = "identity", fill = "#008b8b")barを#008b8b にすることが出来ました。
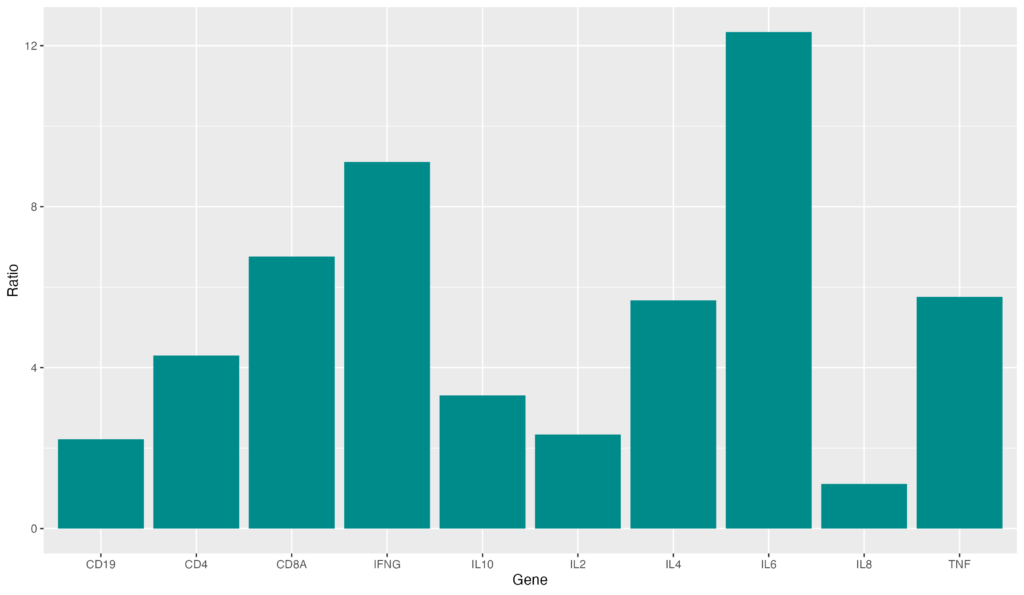
特定のbarの色だけも変更可能です。その場合は以下のようなコードを書きます。
gene_ratio$color[gene_ratio$Gene == "IL6"] <- "red"
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio, fill = color)) +
geom_bar(stat = "identity")IL-6だけの色を変えることが出来ました。一番発現量が高いbarにこのように色を付けるなどするとグラフが見やすく成ると思います。
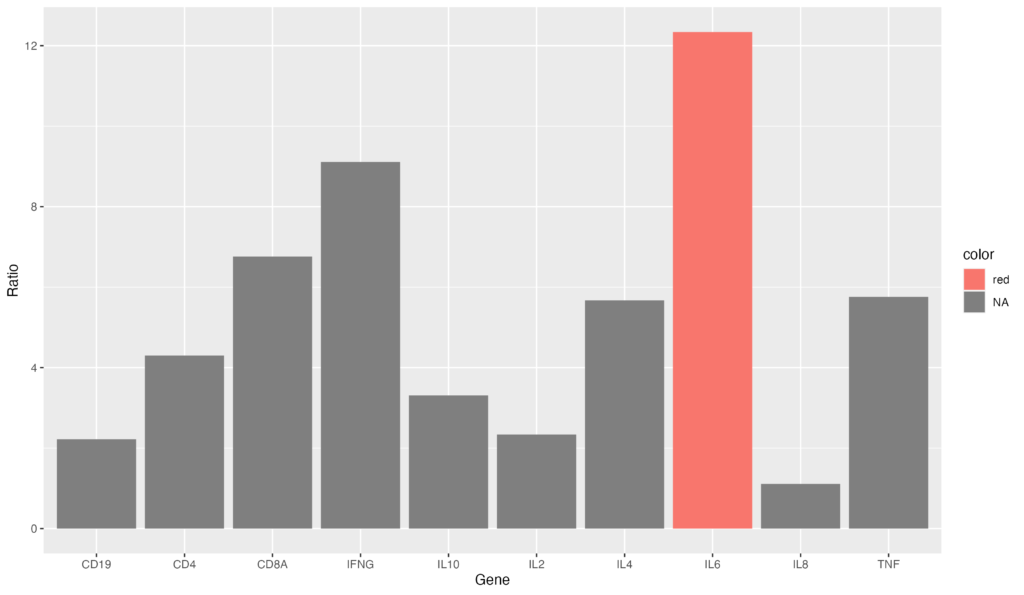
降順にソートするコードと組み合わせれば、最も発現量の大きい遺伝子を図示することも可能でしょう。
gene_ratio$color[gene_ratio$Gene == "IL6"] <- "red"
ggplot(data = gene_ratio, aes(x = reorder(Gene, -Ratio), y = Ratio, fill = color)) +
geom_bar(stat = "identity")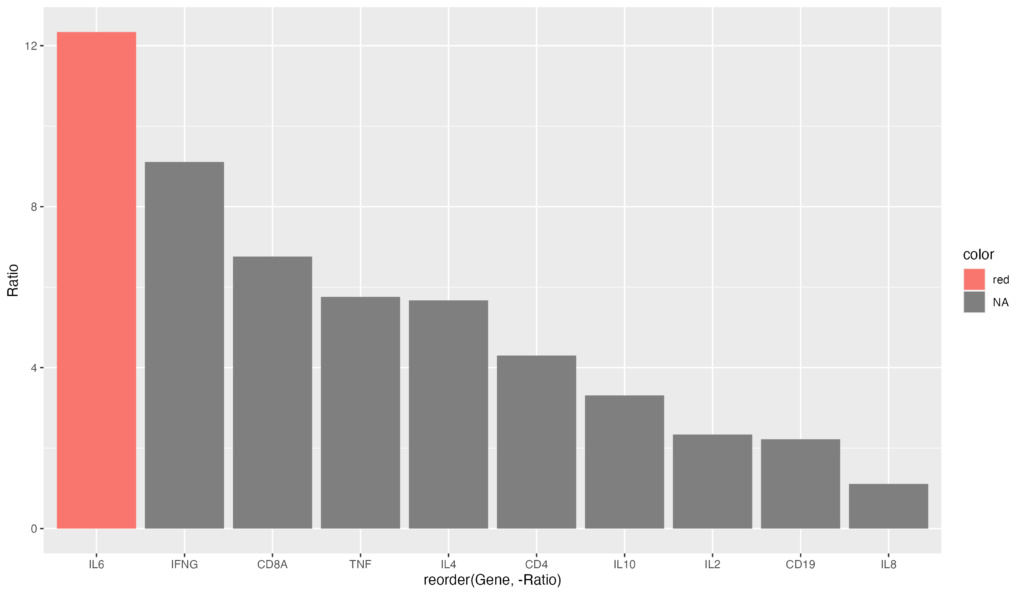
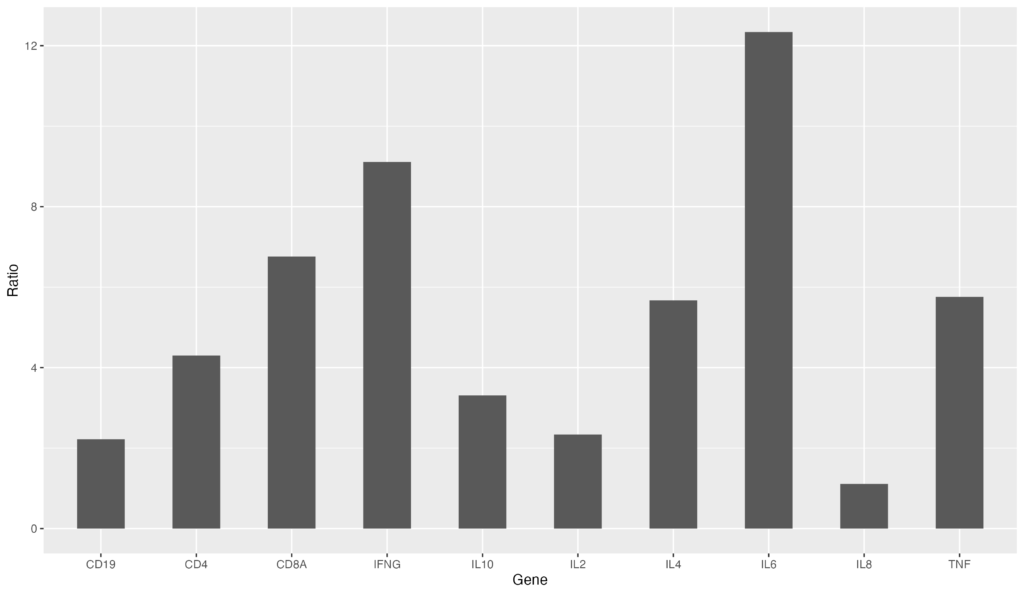
次に、barの幅を調整してみましょう。geom_bar内で、width値を指定します。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity", width=0.5)幅を0.5に変更する事ができました。
論文にグラフを載せたいときにFigureのアスペクト比を調整する必要がある場面は多いと思います。ggplotでは theme(aspect.ratio = 1) のように指定するだけでアスペクト比変更が可能です。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
theme(aspect.ratio = 1)アスペクト比が1で出力出来ました。
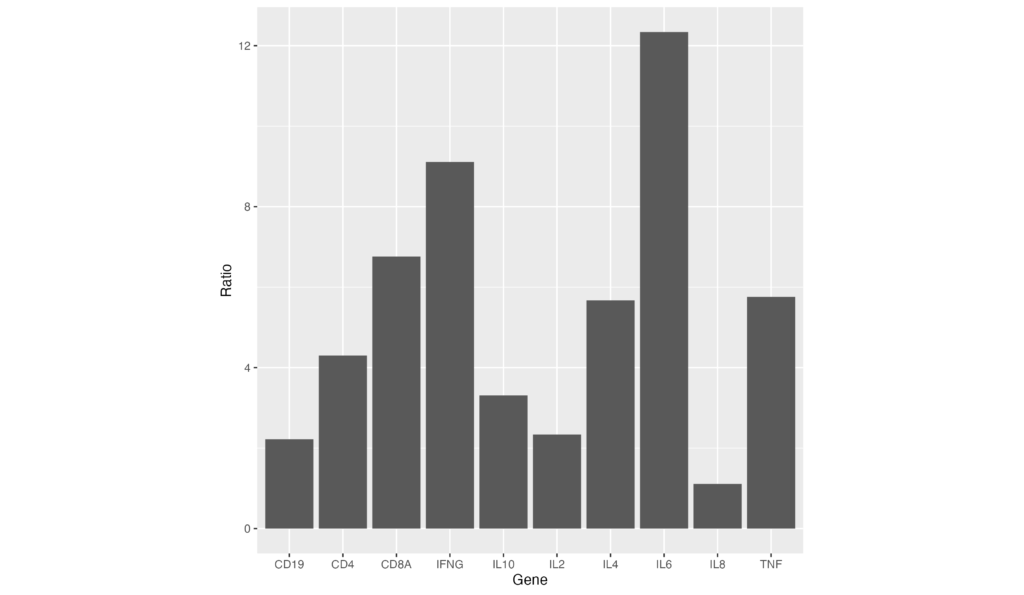
白地の背景にx,y両軸の補助線をかかないようにするには、theme_classic を指定します。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
theme_classic()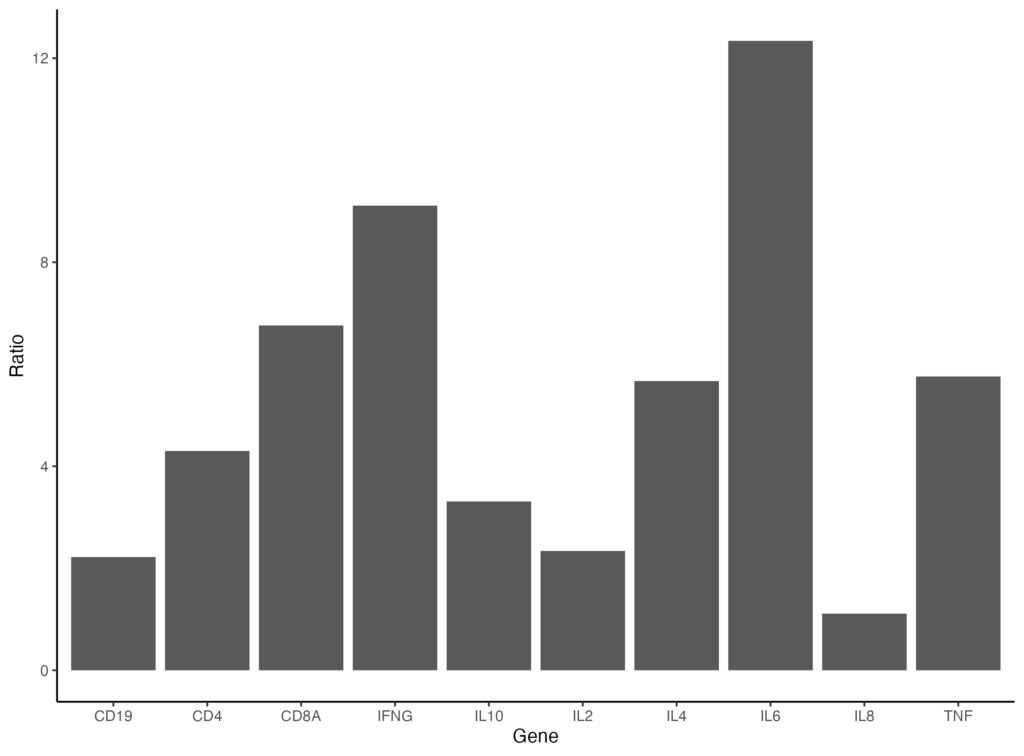
更にプロットエリアの外枠を囲いたいときは、panel.border を以下のように指定します。
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat="identity") +
theme_classic() +
theme(panel.border = element_rect(color = "black", fill = NA))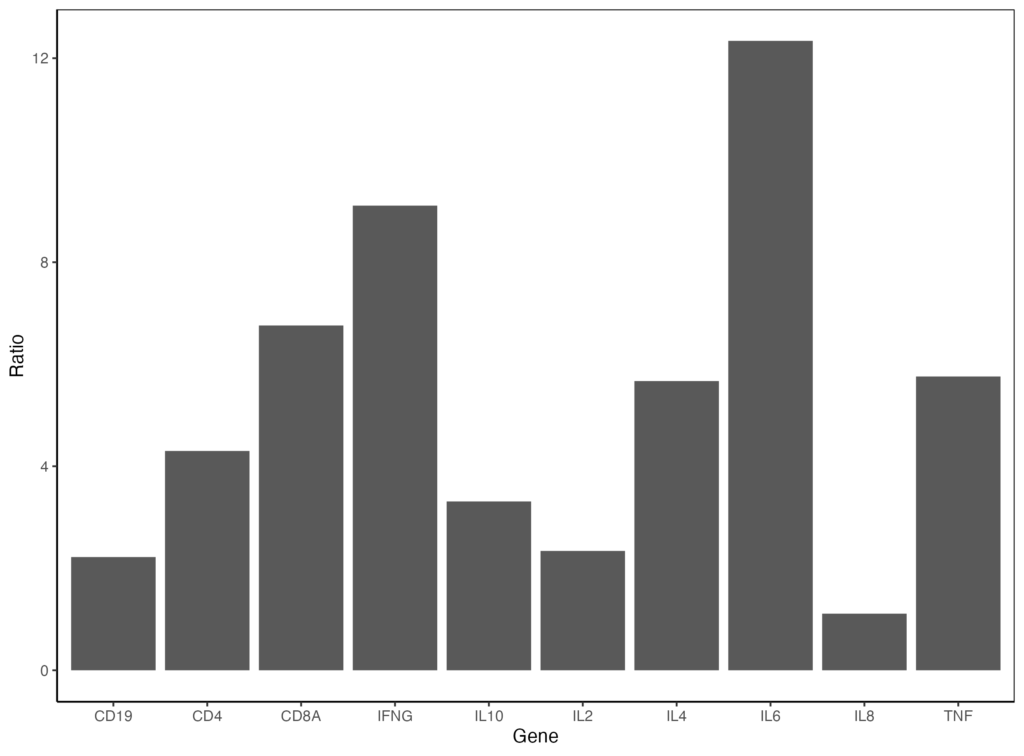
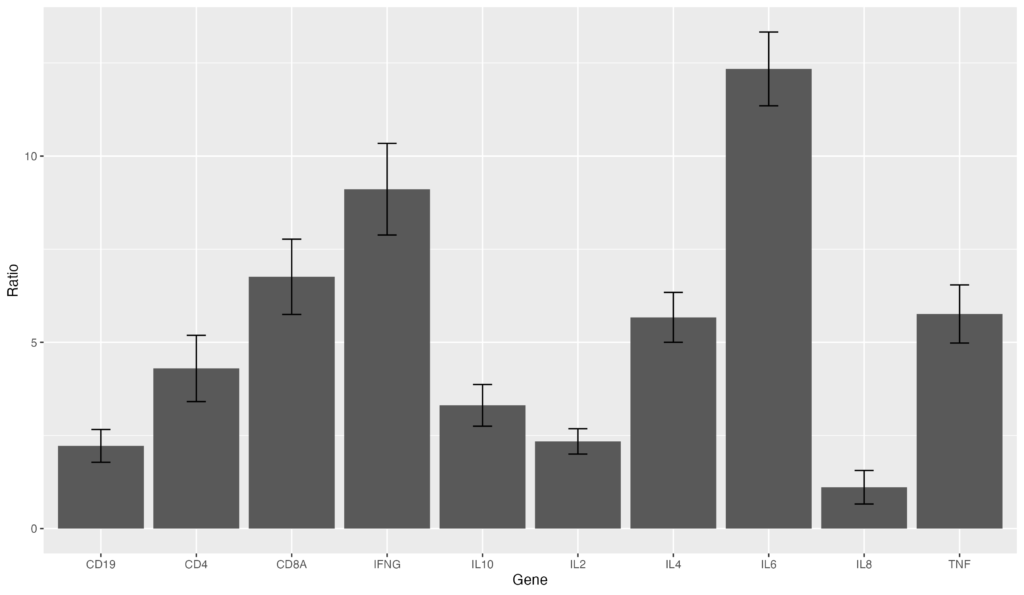
次に、棒グラフにエラーバーを追加してみましょう。エラーバー用の標準偏差をSDとしてgene_ratioに追加してみます。その後、geom_errorbar(aes(ymin = Ratio - SD, ymax = Ratio + SD), width = 0.2)をコードに追記します。
gene_ratio <- data.frame(
Gene = c("IL2", "IL4", "IL6", "IL8", "IL10", "TNF", "IFNG", "CD4", "CD8A", "CD19"),
Ratio = c(2.34, 5.67, 12.34, 1.11, 3.31, 5.76, 9.11, 4.30, 6.76, 2.22),
SD = c(0.34, 0.67, 0.99, 0.45, 0.56, 0.78, 1.23, 0.89, 1.01, 0.44)
)
ggplot(data = gene_ratio, aes(x = Gene, y = Ratio)) +
geom_bar(stat = "identity") +
geom_errorbar(aes(ymin = Ratio - SD, ymax = Ratio + SD), width = 0.2)棒グラフにエラーバーを追加することが出来ました!
最後は、これまで出力してきた図を保存する方法を紹介します。ggsave 関数を使うと画像で保存できます。以下のように保存するファイル名を指定します。
ggsave("sample.png")画像を保存する際、dpiを指定すると、画質を調整可能です。論文の解像度を意識して適切な値を指定しましょう。
ggsave("sample.png", dpi=500)試しに、dpiが100と500の画像を出力してみました。拡大してみると、500のほうがはっきりと文字まで見えるのがわかると思います。
以上、最後にこれまで紹介したコードを組み合わせてグラフを作成してみました。
gene_ratio <- data.frame(
Gene = c("IL2", "IL4", "IL6", "IL8", "IL10", "TNF", "IFNG", "CD4", "CD8A", "CD19"),
Ratio = c(2.34, 5.67, 12.34, 1.11, 3.31, 5.76, 9.11, 4.30, 6.76, 2.22),
SD = c(0.34, 0.67, 0.99, 0.45, 0.56, 0.78, 1.23, 0.89, 1.01, 0.44)
)
gene_ratio$color[gene_ratio$Gene == "IL6"] <- "f08080"
ggplot(data = gene_ratio, aes(x = reorder(Gene, -Ratio), y = Ratio, fill = color)) +
geom_bar(stat = "identity", width = 0.8) +
geom_errorbar(aes(ymin = Ratio - SD, ymax = Ratio + SD), width = 0.2) +
xlab("GeneName") +
labs(title = "Gene Ratio") +
theme_classic() +
theme(plot.title = element_text(hjust = 0.5),
axis.text = element_text(size = 10),
axis.title = element_text(size = 15),
legend.position = "none",
aspect.ratio = 1,
panel.border = element_rect(color = "black", fill = NA))
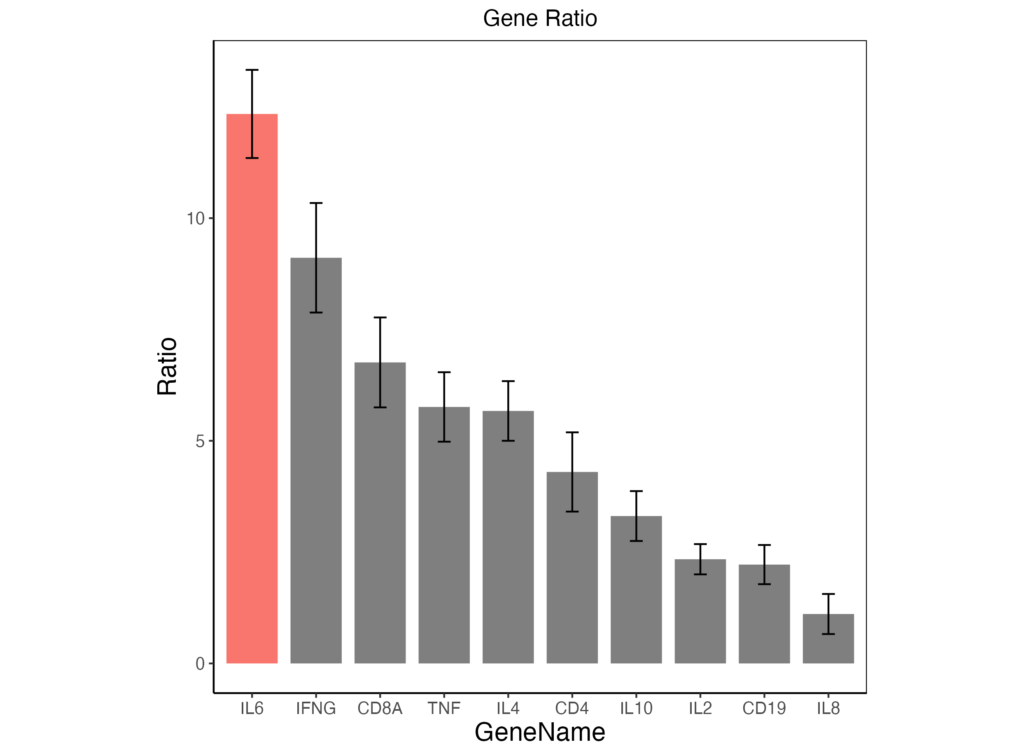
ggsave("sample.png", dpi=500)以上のコードから出力されるグラフは以下のようになります。論文に載せれるレベルのグラフになってきたと思います。
最後に
本記事では、RNA-seq解析に必要なRの知識について、ggplotを例に挙げながら説明しました。ggplotを使えば、棒グラフの作成や軸のフォントサイズの変更、グラフタイトルの追加、縦軸調整、エラーバーの追加などが可能です。ぜひともチャレンジしてください。
公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

